
While the strength, ductility and toughness are most important characteristics for structural materials, there is trade-off relationship between strength and ductility/toughness. A great number of efforts have been devoted to improve the mechanical properties. Controlling "Defect texture" and "alloying" are typical guidelines and empirical approaches have been traditionally used to improve these properties. However, nano- and meso-scopic feature of defect behavior should be understood for more advanced materials design. Here, we have dedicated to develop two approaches: (A) direct modeling and simulations for complicated defect structures, and (B) Non-empirical approaches to evaluate the mechanical properties based on DFT calculations (Fig. 1).
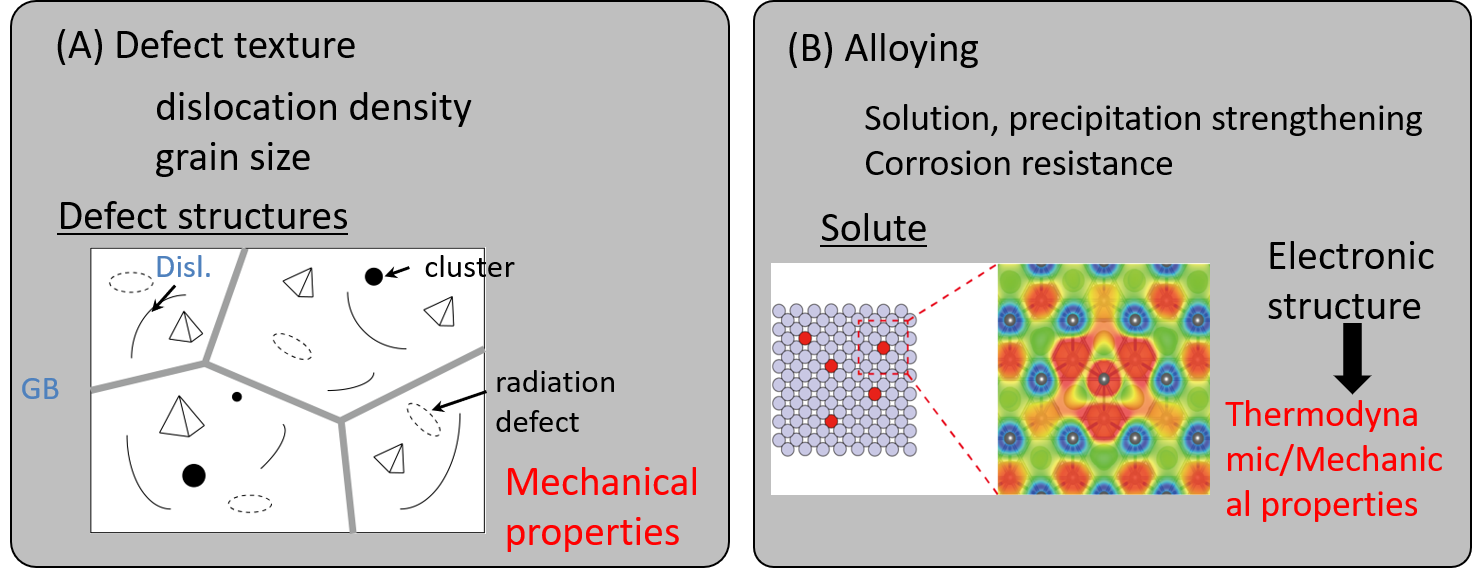
Fig. 1 Schematic images for the improvement of mechanical properties.
(A)We have developed a hybrid parallel atomistic simulation and visualization codes using MPI-IO and applied to the collective defect dynamics of the large-scale atomic models as shown in Fig. 2. Complicated defect dynamics such as defect nucleation and interaction under deformation can be treated directly by atomistic simulations.
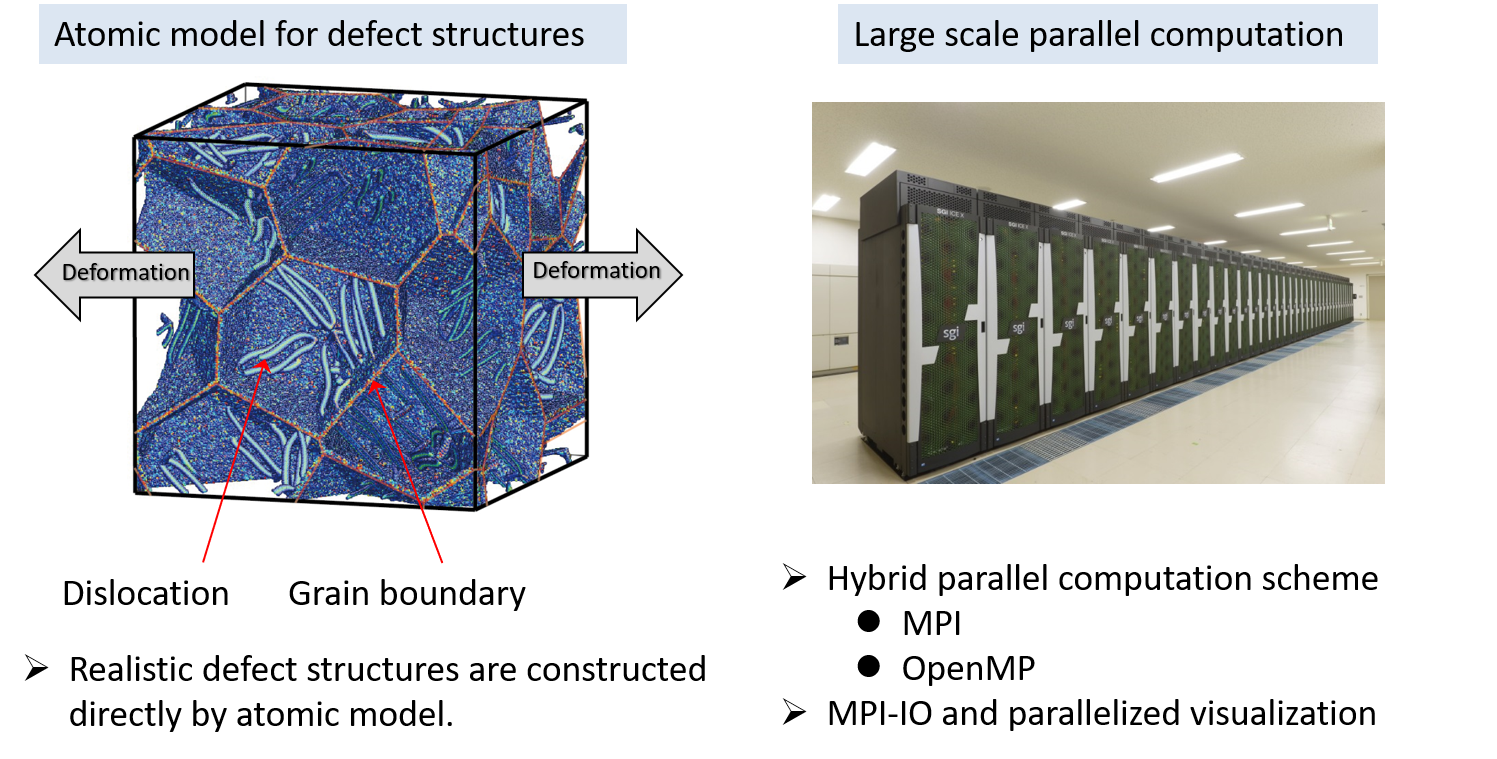
Fig.2 Atomic model for defect structures and parallel computation technique.
(B) We have developed some approaches to evaluate thermodynamic and mechanical properties. Figure 3 shows conceptional images. Here, DFT calculations are employed to evaluate the free energy calculations for thermodynamic properties. The stacking fault energy (SFE) and the energy of dislocation core calculated by DFT are combined with some analytical models to estimate the mechanical properties.

Fig. 3 Theoretical background for mechanical properties derived from DFT calculations.