
Understanding on stress corosion cracking (SCC) and irradiation assisted stress corosion cracking (IASCC) of metalic material is important in tecnological industry. However its mechanism is not clear yet because of the complexity of its phenomena and the difficulty in experiments. Thus, expectations for theoretical investigations at atomic level is rising. We perform theoretical investigations using molecular orbital calculations in order to understand about the influence of impurities on chemical bonds and electronic characterizations of materials that mainly contain iron atoms and the adsorption of various gasses and corrosion mechanism induced on its material surface.
Influences of Ni substitutions to binding energies of Fe6 cluster is shown in Fig. 1. The Ni substitutions decrease the value of binding energy of Fe6 cluster. This suggests that the increase in Ni in grain boundaries of metallic materials, that mainly contain iron, weaken the strength of grain boundaries.
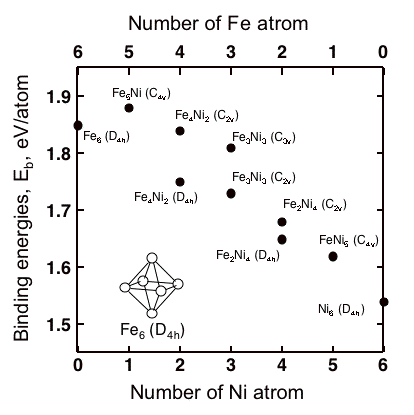
Fig1. Binding energies of Fe6-xNix
We also obtain geometric parameters, binding energies, natural populations, natural electron configurations and magnetic moments for the clusters of Fen, Crn and Fen-xCrx (n = 2-6, x = 1-6) at the UB3LYP/LanL2DZ level. The binding energies of Fen are generally decreased by successive substitutions of Cr for Fe atoms. In the mixed clusters most of Cr-Cr bond lengths are larger than the Fe-Fe and Fe-Cr bond lengths because of the strong repulsion due to the magnetic frustration between atoms.

Embrittlement is known to be caused by P segregation at grain boundaries in Fe alloys. Effects of P substitutions on binding energies and electronic structures of octahedral Fe cluster are investigated using density functional calculations in order to understand the nature of bonding between P and Fe atoms at grain boundaries. The binding energies increase in Fe3P3 and Fe-rich clusters while they decrease in P-rich clusters. The changes in binding energies are closely connected to the charge transfer from Fe to P atoms. The charge transfer leads to both stronger and weaker bonds in mixed Fe-P clusters. The weaker bonds due to less charge cause embrittlement. The calculations indicate that the binding energies and chemical bonding are affected by atomic configurations of P atoms in Fe-P clusters.
The reactions of Fe and FeO2 with O2 and the products of these reactions are investigated at the B3LYP/6-311+G(d) level. The reactions are considered in terms of the calculated potential energy surfaces, the interaction energies between reactant species, and the energies required to populate the higher electronic states such as the excited states of Fe(5F, 3F) and O2(1Σg+). It is found that the reactions of Fe with O2 are endothermic and that the direct formation of dioxide OFeO is due to an ionic interaction of Fe+ with O2–. Furthermore, the diabatic transitions from the covalent and ionic surfaces onto another ionic surface with energy barriers allow the Fe + O2 reaction to proceed toward the formation of OFeO. There are other paths corresponding to the vertical excitation from peroxide Fe(O2) to dioxide OFeO. The OFeO + O2 reactions are found to endothermically produce the η2- and η1-(O2)FeO2 complexes, and the low-lying states of complexes are found to be closely located in energy. The CCSD and CCSD(T) single point energy calculations are performed with the structures optimized at the B3LYP level. The results of NBO and Mulliken analyses and the harmonic vibrational frequencies are presented.